О болести
Фенилкетонурија (ПКУ) је генетска болест која је повезана са метаболичким поремећајем аминокиселина, углавном фенилаланина. Унос обичне протеинске хране у тело бебе доводи до акумулације токсичних метаболичких производа у ткивима. Ова једињења неповратно утичу на дететов нервни систем и мозак, узрокујући прогресивну деменцију.
У неким земљама болест је названа по проналазачу и назива се Воллингова болест. Друго име, фенилпирувична олигофренија, указује на патогенезу болести.
Статистика ширења наследне болести разликује се од земље до земље. Дакле, у Русији се болест налази код једне бебе од 5-10 хиљада новорођенчади (у зависности од региона земље). Најсигурнија земља у погледу ризика од развоја фенилкетонурије је Финска (1: 100 000). Турска је на првом месту у појави генетске болести, једно од 2600 новорођенчади има поремећен метаболизам фенилаланина.
Међу пацијентима са фенилкетонуријом, девојчице су 2 пута вероватније од дечака. Наслеђивање се дешава на аутосомно рецесивни начин, што значи присуство мутирајућег гена у породицама оба родитеља. Здрави носиоци генетског дефекта можда нису свесни сопствених карактеристика и можда немају никакве спољне манифестације. Ситуација се усложњава када се власници мутације венчају и треба да имају децу. Ризик од бебе са фенилкетонуријом у овом случају је 25%. Половина деце рођене у овој породици остаће асимптоматски носиоци наследног оштећења.

Иако болест може довести до грубих, неповратних поремећаја у развоју бебе и значајно погоршати квалитет живота бебе, непријатне последице могу се избећи. Фенилкетонурија је једна од ретких наследних болести која се може ефикасно лечити. Уз благовремене мере, беба готово увек расте здраво.
Историјска референца
По први пут је на наследну природу деменције сумњао норвешки биохемичар и психијатар Ивар Асбјорн Фоллинг. Посматрајући групу ментално заостале деце, лекар је уочио заједничку клиничку слику и утврдио промену хемијског састава урина болесне браће и сестара. Додајући раствор жељезног хлорида у биолошку течност, лекар је открио промену боје урина, појаву маслинасто-зелене нијансе. Присуство промена у анализама код блиских рођака изазвало је сумњу у наследне узроке болести.
Овај метод откривања фенилкетонурије није изгубио своју важност у савременом свету. Одређивање фенилпирувичне киселине у урину назива се Воллингов тест у част норвешког лекара.

Како се болест развија?
Немогуће је предвидети развој фенилкетонурије код новорођене бебе без додатних истраживања. Беба се не разликује од својих вршњака, изгледа као апсолутно здраво дете. Промене се дешавају у телу када започне ентерално храњење, када протеини мајчиног млека уђу у тело.
Узрок патологије лежи у недостатку одређених ензима који учествују у конверзији есенцијалне аминокиселине фенилаланина у тирозин. Тако се повећава ниво фенилаланина и његових деривата у крви и цереброспиналној течности. Метаболички производи имају токсични ефекат на мождане ћелије, ремете метаболизам масти и пренос нервних импулса између неурона.
Истовремено, тирозин, неопходан за нормално функционисање, не синтетише се у одговарајућој количини. Ова аминокиселина је неопходна и активно учествује у стварању хормона, неуротрансмитера, пигмента меланина.
Постоји неколико клиничких облика фенилкетонурије, које карактерише недостатак одређеног ензима у јетри. У 98% се јавља ПКУ тип И, повезан са дефектом гена у 12. хромозому. Болест се манифестује недостатком фенилаланин-4-хидроксилазе и добро реагује на лечење. ИИ и ИИИ врсте болести су изузетно ретке болести и одликују се тешким манифестацијама, неефикасношћу дијететске терапије у корекцији метаболизма аминокиселина.
Симптоми класичног облика фенилкетонурије
Уобичајене манифестације
Ако у породилишту није извршен неонатални скрининг, болест је остала непрепозната и нису организоване потребне мере лечења, тада ће се ПКУ манифестовати у првој години бебиног живота. Родитељи ће прве знаке одступања од норме пронаћи код детета у доби од 2-6 месеци, када претходно здраво дете проводи летаргичност и апатичност, или обрнуто, претерано раздражљиво. Једење хране често се завршава регургитацијом, а на бебиној кожи појављују се манифестације алергијског дерматитиса.
Карактеристичан изглед
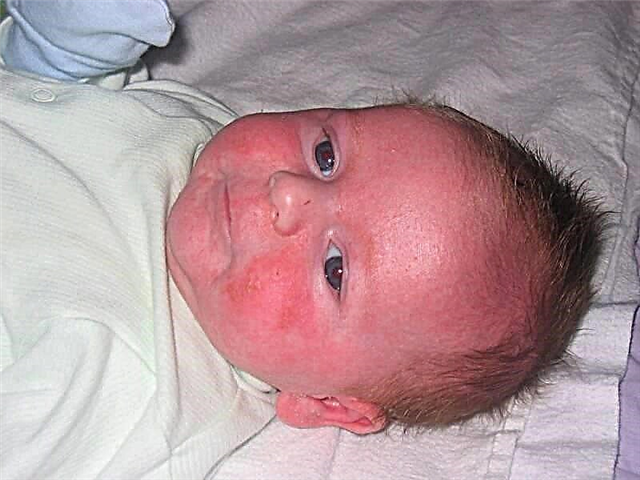
Пошто кршење метаболизма аминокиселина доводи до смањења стварања меланина, у случају развоја болести, кожа, коса и ирис очију губе пигмент. Бебе са генетским поремећајем имају плаву косу, танку бледу кожу и плаве очи. Карактеристичан мирис зноја и урина мрвица може указивати на наследну болест. Многи лекари га описују као „миша“, а повезан је са ослобађањем метаболичких производа аминокиселина са биолошким течностима.

Оштећење нервног система
Први знаци промена у нервном систему манифестују се летаргијом, смањењем мишићног тонуса мрвица. Често се јавља дистонија, појављују се нехотичне контракције мишића, опсесивни покрети, конвулзије. Могућ је развој органских патологија мозга - микроцефалија и хидроцефалус.
Скоро 50% пацијената са фенилкетонуријом развија епилепсију. Понекад су карактеристични напади први знак наследне болести. Пароксизме је тешко лечити конвенционалним антиконвулзивима и значајно смањују квалитет живота детета.
Заостајање у развоју
Са током болести, беба брзо губи интересовање за свет око себе, постаје апатична. Физички развој детета такође пати - беба касно савлада потребне вештине. Кашњење у менталном развоју постаје очигледно када се појаве проблеми у формирању говора, заостајање у менталном развоју.

Студије су показале да се грубе промене у интелектуалној активности развијају након годину дана прогресије болести. Мрвица неповратно губи до 50 ИК бодова годишње, што последично доводи до развоја ослабљености, имбецилије или идиотизма. Стога је време почетка лечења фенилкетонурије веома важно.
Неуролошки проблеми код старије деце изражавају се у хиперактивности, појави стереотипа, опсесивним покретима руку, лица, језика, њихању. Понекад се ментални поремећаји манифестују у шизофреним стањима.
Озбиљно кашњење у развоју доводи до недостатка вештина ходања код 30% болесне деце и алалије, неразвијености говора, код 60% пацијената.
Разноликости патологије
Прелазни облик хиперфенилаланинемије
Болест се не одвија увек класично, понекад је пораст нивоа фенилаланина привремен, а сама болест се не развија. Краткорочно повећање концентрације аминокиселина у крви детета може бити повезано са незрелошћу ензимских система бебе, што је најтипичније за дубоко превремено рођену бебу. Такве бебе захтевају детаљнији преглед како би се утврдио узрок болести.
Птерин зависне варијанте фенилкетонурије
Отприлике 2% беба којима је дијагностикована фенилкетонурија у породилишту и којима је прописана одговарајућа дијета има неуролошке проблеме. Ови симптоми могу указивати на развој ретког, птерин зависног облика ПКУ (тип ИИ, ИИИ према класификацији) код бебе. Клиничке манифестације атипичних облика ПКУ сличне су току класичног облика, али симптоми брже напредују, а дијететска терапија нема жељени ефекат.

Огромну улогу у развоју облика који зависе од птерина игра недостатак неуротрансмитера, који се јавља услед кршења метаболизма аминокиселина. Ове супстанце су неопходне за пренос нервних импулса између нервних ћелија, без којих је нормално функционисање тела немогуће.
Фенилкетонурија ИИ се сматра малигнијом од типа ИИИ. Груби метаболички поремећаји аминокиселина доводе до масовне смрти нервних ћелија. Бебе са прогресивним ПКУ типа ИИ често умиру у доби од 2 до 3 године.
Мајчинска фенилкетонурија
Деца са ПКУ нису увек рођена од здравих родитеља. Ако труднице са наследним поремећајима не следе дијету, постоји велика вероватноћа да ће имати дете са синдромом фенилкетонурије по мајци. Токсичне супстанце продиру кроз плаценту од мајчина тела до фетуса и нарушавају стварање дечјих органа и система. Деца се рађају са вишеструким урођеним малформацијама, микроцефалијом, менталном и физичком ретардацијом.
Дијагностика
Породично генетско саветовање
Будући родитељи морају бити посебно опрезни ако је у породицама било ког од њих било случајева рођења деце са фенилкетонуријом. Не може се искључити могућност рођења детета са ПКУ, чак и ако се болест манифестовала у даљем рођаку. Подмуклост болести лежи у асимптоматском ношењу оштећеног гена од стране апсолутно здравих људи.
Сви парови који у породици сумњају на наследне болести треба да се обрате генетичару чак и током планирања бебе. У Медицинско-генетском центру (МГЦ), користећи савремене дијагностичке методе, могуће је идентификовати ношење мутантног гена и израчунати ризик од рађања детета са ПКУ и другим генетским болестима.
Инвазивне дијагностичке методе (хорионска биопсија, амниоцентеза, кордоцентеза) могу помоћи у утврђивању болести пре рођења бебе. У овом случају се испитује генетски материјал добијен од фетуса. Ове методе су трауматичне и могу довести до спонтаног побачаја. Стога је њихова употреба оправдана само доказаним ношењем мутираних гена код родитеља и великим ризиком од ПКУ код бебе.
Неонатални скрининг

Пре отпуштања из породилишта, новорођене бебе се масовно испитују на наследне болести. За ову важну студију, здравствени радник вади крв из бебине пете и наноси капљице биолошке течности на део филтера тестне плочице. Свака кап крви је дизајнирана за откривање једне од 5 болести: фенилкетонурија, цистична фиброза, урођени хипотироидизам, адреногенитални синдром, галактоземија.
Неонатални скрининг је веома важан и неопходан преглед за сву децу без изузетка. Спроводи се потпуно бесплатно и има за циљ очување здравља нације. Одбијањем манипулације, родитељи праве огромну грешку, јер симптоми наследних тегоба нису увек приметни код новорођене бебе. Живописне клиничке манифестације често настају када је детету већ тешко помоћи, а промене у телу мрвица постале су неповратне.
Ова метода је поуздана у откривању фенилкетонурије, ако беба прима довољну количину ентералне исхране, мајчино млеко. Стога се новорођенчад која се налазе на одељењу интензивне неге анализира касније. Распоред испитивања код доношене и недоношчади такође се разликује. За бебе које су се појавиле на време, препоручује се сет капиларне крви 4. дана живота. Студија о превремено рођеним мрвицама одлаже се за 7 дана.
Анализу треба извршити на празан стомак, што ће пружити тачнији резултат и повећати дијагностичку вредност теста. Образац са капљицама крви бебе и подацима о пасошу родитеља шаље се у лабораторију медицинско-генетске консултације, где се спроводи биохемијска студија.
Родитељи треба да обрате пажњу на тачност попуњавања формулара, тачност својих података за контакт. Студија траје у просеку 10 дана, а породица је у то време обично код куће. Ако се утврди позитиван резултат теста, родитељи ће морати да контактирају медицински генетски центар што је пре могуће, а нетачни подаци за контакт одложиће даљи преглед бебе.
Преглед детета у Московском центру града
Да би потврдила болест код новорођенчета и утврдила узрок, беба ће морати да прође поступак у више фаза. Поновљени биохемијски тестови помоћи ће да се потврди пораст нивоа аминокиселина у крви и донесе одлука о потреби за дијеталном терапијом. Извршава се студија нивоа фенилаланина и тирозина, активности ензима јетре, одређивање метаболичких производа аминокиселина у урину.
Затим се врши молекуларно-генетска дијагностика, што омогућава идентификацију мутације у гену РАС одговорног за развој фенилкетонурије. Користећи ове методе, могуће је утврдити асимптоматски пренос болести.
Третман ПКУ
Најефикаснијом методом лечења класичног облика фенилкетонурије сматра се правилно организована дијетална терапија. Само искључивањем хране која садржи велику количину аминокиселина из исхране мрвица можемо очекивати позитивне резултате лечења.
Карактеристике дијететске терапије
Правила избора хране
Фенилкетонурија код деце је тешка болест и само лекар може одабрати праву дијету за бебу. Приликом избора дозвољених производа и израчунавања њихове запремине, лекар узима у обзир неке појединачне карактеристике:
- који је облик болести присутан код бебе;
- тежина болести, ниво фенилаланина у крви;
- старост мрвица;
- особине физичког развоја врпоља;
- физиолошке потребе организма који расте за протеинима;
- како беба толерише унос мале количине фенилаланина из хране.
Аминокиселина фенилаланин је незамењива, што значи да се може уносити само са протеинском храном. Растућем детету је потребна довољна количина хранљивих састојака, јер се око 40% фенијаланина у исхрани троши на изградњу властитих протеина. Испоставља се да је такође немогуће потпуно искључити опасне производе из исхране мрвица. Због тога се првих дана дијете терапије у лабораторији процењује како тело мрвица реагује на мали унос аминокиселина са храном.
Да ли је дојење могуће фенилкетонуријом?
Пошто се у већини случајева болест утврђује у првим недељама бебиног живота, поставља се питање сигурности храњења бебе мајчиним млеком. У ствари, дојење је неопходно за нормалан развој детета, формирање његовог имунолошког система. Али његова употреба је оправдана само у случајевима када се мајка придржава јасних правила и строго контролише количину хране коју беба поједе.
За сигурно храњење, боље је користити свеже издојено мајчино млеко, чија дозвољена количина лекар израчунава на основу телесне тежине бебе и његове потребе за фенилаланином.
Што је дете млађе, већа му је потреба за аминокиселинама. На пример, дозвољена количина фенилаланина за новорођену бебу је 90 - 60 мг / кг телесне тежине дневно, док је потребна количина аминокиселина за школарца смањена на 10 - 20 мг / кг телесне тежине. Количина фенилаланина у храни може се израчунати с обзиром на то да 1 г протеина садржи око 50 мг аминокиселине.
Специјализоване смеше
Постоје посебно развијени производи за рационалну исхрану деце са фенилкетонуријом. Направљени су на бази протеинских хидролизата или смеше аминокиселина, који су у потпуности или делимично лишени фенилаланина. Прехрана лековитим смешама има за циљ допуњавање тела потребним протеинима, довољном залихом хранљивих састојака. Ове формуле се могу користити као замена за мајчино млеко или као додатак главној исхрани за старију децу.
Састав лековитих смеша за децу различите старости је различит. На пример, за децу млађу од једне године погодни су такви производи као што су "Афенилак", "Лофеналак". Старијој деци су приказани "Тетрафен", "Без фенила", "Макамум - КСП".

"Семафор за храну"
Сваке године беба расте, а потребе за храном се повећавају. Време је да уведете комплементарну храну, испробајте нова јела. Родитељи морају да се сете опасности фенилаланина за бебин мозак, бирају само храну коју је одобрио лекар.
Исхрана болесног детета значајно се разликује од исхране вршњака. Допунско храњење започиње увођењем поврћа и воћа, затим треба додати тестенине без протеина, неке врсте житарица. Лекар који треба да састави бебин први мени и контролише садржај аминокиселина у крви у позадини промена у исхрани.
Сви производи су подељени у групе према садржају фенилаланина у њима:
- црвени списак.
Препоручује се да храну са ове листе у потпуности избаците јер је богата протеинима. То су месо и остаци, риба, јаја, хлеб, хељда, јечмене житарице, Херкулове пахуљице;
- жута листа.
Дозвољено је користити малу количину производа из ове групе под контролом теста крви. Можда увођење кукурузне и пиринчане крупице, кромпира, млечних производа са малим садржајем протеина;
- зелена листа.
Ови производи чине основу исхране старије деце са фенилкетонуријом: кукурузно и пиринчано брашно, већина поврћа, воћа и бобица, слаткиши, путер.
Када користите готове производе, морате пажљиво прочитати упутства на паковању. Једење банана, сувог воћа и махунарки може повећати концентрацију фенилаланина, па их треба ограничити.

Остале терапије
Употреба лекова је оправдана само за лечење облика ПКУ зависних од птерина, када дијета нема жељени ефекат. У таквим ситуацијама користе се лекови Л-допа и 5-хидрокситриптофан.
У другим случајевима је могуће користити витаминску терапију, минералне комплексе, као додатак исхрани. Ако је потребно, прописани су ноотропни лекови, антиконвулзиви. Рад са логопедима, психолозима, масажом, физиотерапијом такође има позитиван резултат у лечењу болести.
Прогноза и превенција
Ток класичног облика болести не утиче значајно на очекивани животни век особе. Али без правилног лечења, тешки симптоми болести значајно утичу на интелигенцију и квалитет живота пацијента. У случају рационалне дијететске терапије и правовремене контроле нивоа фенилаланина у крви, деца са ПКУ се не разликују од својих вршњака и добро се прилагођавају у друштву. Атипични облици болести често доводе до инвалидитета и смрти детета, јер лечење ретко даје добре резултате.
Стручњаци препоручују да се дијете придржава током читавог живота, али је могуће олакшање када дете напуни 18 година. Посебну пажњу треба посветити трудницама са ПКУ како би се избегао синдром фенилкетонурије мајке код детета.
Превенција се састоји у спречавању блиско повезаних бракова, медицинском и генетском саветовању породица у ризику за развој ПКУ и масовном скринингу новорођенчади.

Закључци
Бебина болест представља велики стрес и тест за младе родитеље, посебно ако је болест наследна. Али фенилкетонурија је једна од ретких генетских болести за коју интервенције у лечењу доносе добре резултате.
Родитељи морају да схвате важност скрининга новорођенчади и ране, добро одабране дијететске терапије. Вреди пажљиво пратити све препоруке лекара који присуствује и тада таква страшна дијагноза као фенилкетонурија неће постати реченица за дете.